Lutetium-177 Gastrin-Releasing Peptide Receptor
LY4257496
Phase
1
Recruiting
A Phase 1a/b Multicenter, Open-Label Trial to Evaluate Safety, Tolerability, and Dosimetry of LY4257496, a GRPR-Targeted Radioligand Therapy, in Adults With GRPR-Positive Advanced Solid Tumorsa
Related Resources:
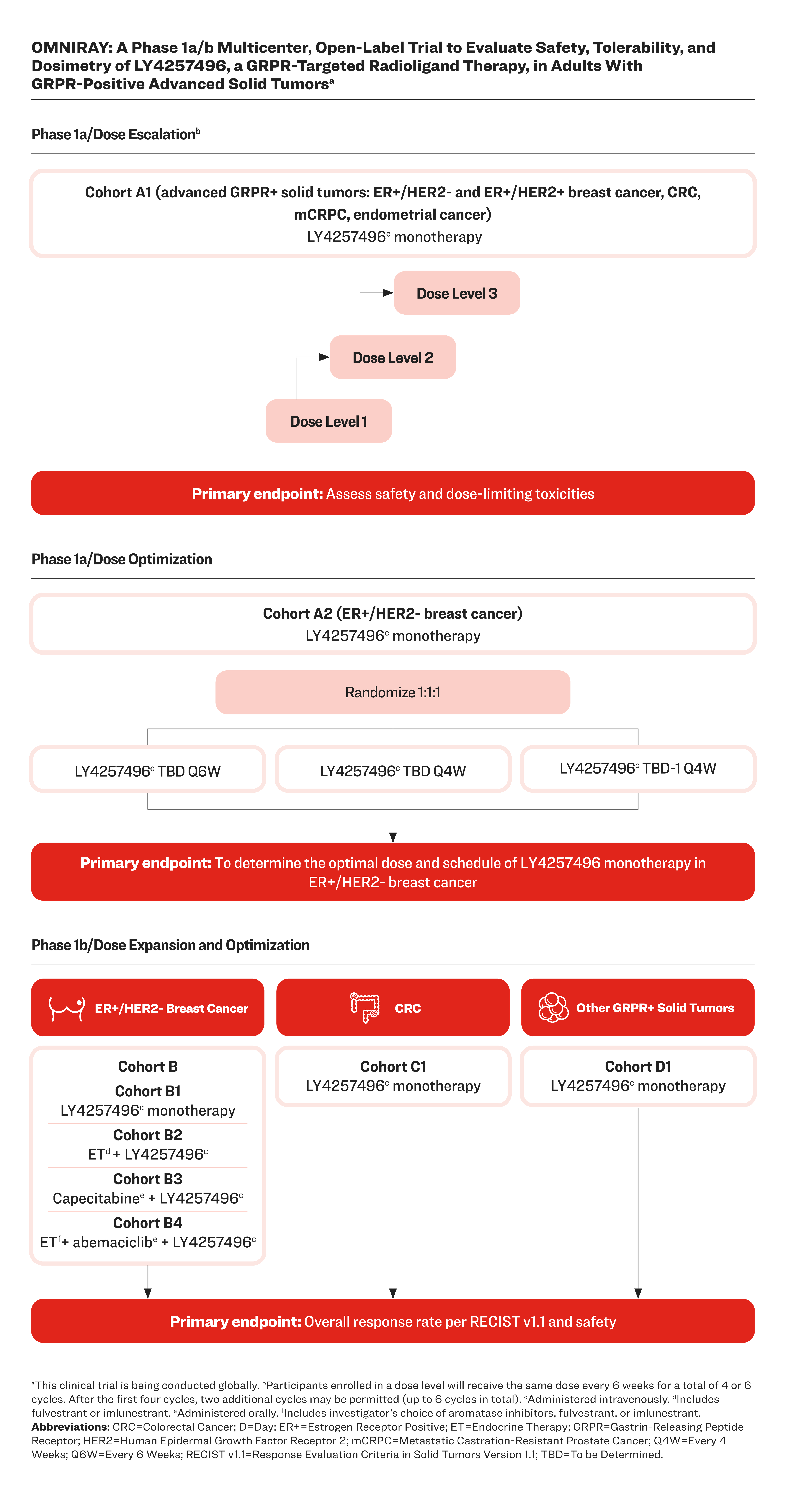
Key Inclusion Criteria
- Male or female adults (≥18 years of age)
- Must have histologically or cytologically proven diagnosis of locally advanced, unresectable, or metastatic cancer
- Must be assessed by computed tomography (CT) scan/magnetic resonance imaging (MRI) to confirm at least 1 of the following:
- At least 1 measurable target lesion per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1
- If only bone lesions are present without a soft-tissue component, a bone scan or MRI must confirm at least 2 detectable lesions considered to represent active metastases
- Must have gastrin-releasing peptide receptor (GRPR)-positive disease, defined by investigator assessment of GRPR imaging
- Must have the following histologically or cytologically confirmed diagnosis:
- Estrogen receptor positive (ER+)/human epidermal growth factor receptor 2 negative (HER2-) breast cancer
- ER+/HER2 positive (HER2+) breast cancer
- Colorectal carcinoma
- Metastatic castration-resistant prostate cancer (mCRPC)
- Endometrial carcinoma
- Other GRPR-positive solid tumor
- Eastern Cooperative Oncology Group (ECOG) performance status ≤1
- Must have adequate organ function:
- Absolute neutrophil count (ANC) ≥1.5 × 109/L
- Platelets ≥100 × 109/L
- Hemoglobin ≥8 g/dL (4.97 mmol/L)
- Total bilirubin ≤1.5× upper limit of normal (ULN)
- Alanine aminotransferase (ALT) and aspartate aminotransferase (AST), ≤3× ULN or ≤5× ULN for participants with documented liver involvement, respectively
- Creatinine clearance (CrCl) ≥60 mL/minute
- Cohort A1:
- ER+/HER2- and ER+/HER2+ breast cancer: Must have received no more than 5 prior lines of therapy for advanced or metastatic disease
- Prior treatment with a CDK4/6 inhibitor (CDK4/6i) is required
- HER2+: At least 1 HER2-directed therapy for advanced disease is required
- Colorectal carcinoma:
- Must have received 1 or more prior fluoropyrimidine-based chemotherapy for advanced or metastatic disease
- Approved therapies are no longer effective or are not considered appropriate or safe in the opinion of the investigator
- Adenocarcinoma of the prostate:
- Castrate circulating testosterone levels (<1.7 nmol/L or <50 ng/dL)
- Must have received no more than 3 prior lines of therapy for advanced or metastatic disease and meet the following criteria:
- At least 1 prior androgen receptor pathway inhibitor (ARPI) (eg, abiraterone, apalutamide, darolutamide, enzalutamide), AND
- At least 1 prior taxane-based chemotherapy for castration-resistant prostate cancer (CRPC) (or was deemed ineligible for or refused taxane-based chemotherapy);
- Prior 177Lu-PSMA-617 and/or poly ADP ribose polymerase inhibitors (PARPi) are permitted
- Neuroendocrine carcinoma of the prostate and endometrial carcinoma:
- Must have received 1 or more prior platinum-based chemotherapy for advanced or metastatic disease
- Cohort A2:
- Prior treatments required:
- ER+ and HER2- breast cancer: Must have received no more than 5 prior lines of therapy in the advanced or metastatic setting, and must have received a prior CDK4/6i
- Prior treatments required:
- Cohort B:
- ER+ and HER2- breast cancer:
- Cohort B1, B2, B3: Must have received no more than 5 prior lines of therapy in the advanced or metastatic setting, and must have received a prior CDK4/6i
- Cohort B4: Must have received no more than 2 prior lines of therapy for advanced disease and may be:
- CDK4/6i naïve, or
- CDK4/6i treatment ongoing, without evidence of progression on or after a CDK4/6i-containing regimen (eg, participants who have already initiated first or second-line treatment with CDK4/6i-containing therapy), OR
- CDK4/6i pretreated, with evidence of progression on or after CDK4/6i-containing therapy
- ER+ and HER2- breast cancer:
- Cohort C:
- Colorectal carcinoma:
- Cohort C1: Must have received prior fluoropyrimidine-containing therapy
- Colorectal carcinoma:
- Cohort D:
- GRPR-positive solid tumor other than ER+/HER2- breast cancer and colorectal cancer:
- Cohort D1: Must have received 1 or more prior line(s) of systemic therapy in the advanced or metastatic setting
- GRPR-positive solid tumor other than ER+/HER2- breast cancer and colorectal cancer:
Key Exclusion Criteria
- Received any radiopharmaceutical or radioligand therapy (eg, Radium-223, Strontium-89, Samarium-153, any [Lu-177]-compounds, any [Ac-225]-compounds, any [Pb-212 compounds). For participants with mCRPC, prior 177Lu-PSMA-617 is permitted
- Received any prior hemi-body or whole-body radiotherapy, or prior external beam radiation therapy (EBRT) to greater than 25% of the bone marrow
- Has a history of or ongoing acute pancreatitis within 1 year of screening
- Has evidence of ongoing and untreated urinary tract obstruction (ie, hydronephrosis or hydroureter) or unmanageable urinary incontinence
- Phase 1a (Cohort A1) only: Currently receiving neprilysin (NEP) inhibitors (ie, Entreso, racecadotril) and for whom images for dosimetry assessments cannot be acquired
- Has known active central nervous system (CNS) metastases and/or carcinomatous meningitis. Individuals with previously treated CNS metastases may participate provided:
- Any prior CNS-directed therapy is completed, including radiation or surgery, ≥14 days prior to the first dose of study intervention
- CNS disease is neurologically and radiologically stable, defined as without evidence of progression for ≥14 days by repeat imaging obtained prior to the first dose of study intervention
- Individuals receiving glucocorticoid therapy must not receive >10 mg prednisone daily or equivalent
- Individuals receiving prophylactic anticonvulsants must be on a stable dose for ≥14 days prior to the first dose of study intervention. Enzyme inducing anti-epileptic drugs for CNS metastases must be discontinued for at least 5 half-lives prior to the first dose of study intervention
- Has any unresolved toxicities from prior therapy greater than National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) (Version 5.0) Grade 1 at the time of starting trial treatment except for alopecia and peripheral neuropathy
- Has significant cardiovascular disease
- Unstable angina or acute coronary syndrome within the past 2 months
- History of myocardial infarction within 6 months prior to planned start of trial treatment
- Documented left ventricular ejection fraction (LVEF) by any method of ≤40% in the 12 months prior to planned start of trial treatment, unless subsequent measurements (≥2 of any kind, separated by a minimum of 3 weeks) document LVEF recovery to >40%
- Grade ≥3 New York Heart Association functional classification system of heart failure, uncontrolled or symptomatic arrhythmias
- Has prolongation of the corrected QTcF >470 msec during screening
- Has active uncontrolled systemic bacterial, viral, fungal, or parasitic infection (except for fungal nail infection), or other clinically significant active disease process which, in the opinion of the investigator and the sponsor, makes it undesirable for the individual to participate in the trial. Screening for chronic conditions is not required
- Has known active hepatitis B virus (HBV) (screening for HBV is not required for individuals who do not have a history of HBV, unless required by local regulations). Individuals with treated/chronic HBV are eligible for the trial provided they meet the following criteria:
- Individuals with positive hepatitis B surface antigen (HBsAg) must be on permitted suppressive antiviral therapy prior to Cycle 1 Day 1 (C1D1), remain on the same antiviral treatment throughout trial, and should follow local standards for continuation of therapy after completion of trial therapy
- Undetectable HBV DNA ≤28 days of C1D1
- Has known active hepatitis C virus (HCV) (screening for HCV is not required for individuals who do not have a history of HCV unless required by local regulations). Individuals previously treated for HCV are eligible for the trial provided they meet the following criteria:
- Completion of curative antiviral therapy
- HCV viral load below the limit of quantification ≤28 days of C1D1
- Negative hepatitis C antibody result OR, if positive, then must be hepatitis C RNA negative
- Has known untreated human immunodeficiency virus (HIV) infection (screening for HIV is not required unless required by local regulations). Participants on permitted antiretroviral therapy (ART) and who have well-controlled HIV infection/disease are eligible provided they meet the following criteria:
- Must be on a stable and permitted ART regimen without changes in drug or dose, for at least 4 weeks prior to C1D1 and have a viral load of <400 copies per mL prior to ≤28 days of C1D1
- Cluster of differentiation 4 positive (CD4+) T-cell count ≥350 cells/uL ≤28 days of C1D1
- Has an active second malignancy unless in remission with life expectancy >2 years
- Has received a vaccination with a live vaccine ≤7 days prior to enrollment
- Is pregnant and/or planning to breastfeed during the trial or within 6 months of the last dose of trial intervention
- Has known hypersensitivity to any component or excipient of LY4257496
- Prior therapies:
- Bisphosphonates or RANK-L targeted agents (eg, denosumab) ≤7 days
- Cytotoxic therapies or targeted agents that are small molecule inhibitors ≤14 days or ≤5 half-lives, whichever is shorter
- Therapeutic monoclonal antibodies ≤28 days
- Radiotherapy
- Palliative limited field radiation ≤7 days
- Major surgery ≤28 days
a
This clinical trial is being conducted globally.
b
Participants enrolled in a dose level will receive the same dose every 6 weeks for a total of 4 or 6 cycles. After the first four cycles, two additional cycles may be permitted (up to 6 cycles in total).
c
Administered intravenously.
d
Includes fulvestrant or imlunestrant.
e
Administered orally.
f
Includes investigator’s choice of aromatase inhibitors, fulvestrant, or imlunestrant.

