Gastrin-Releasing Peptide Receptor Radioligand
LY4257496
Phase
1
Recruiting
A Phase 1a/b Multicenter, Open-Label Trial to Evaluate Safety, Tolerability, and Dosimetry of LY4257496, a GRPR-Targeted Radioligand Therapy, in Adults With GRPR-Positive Advanced Solid Tumorsa
Related Resources:
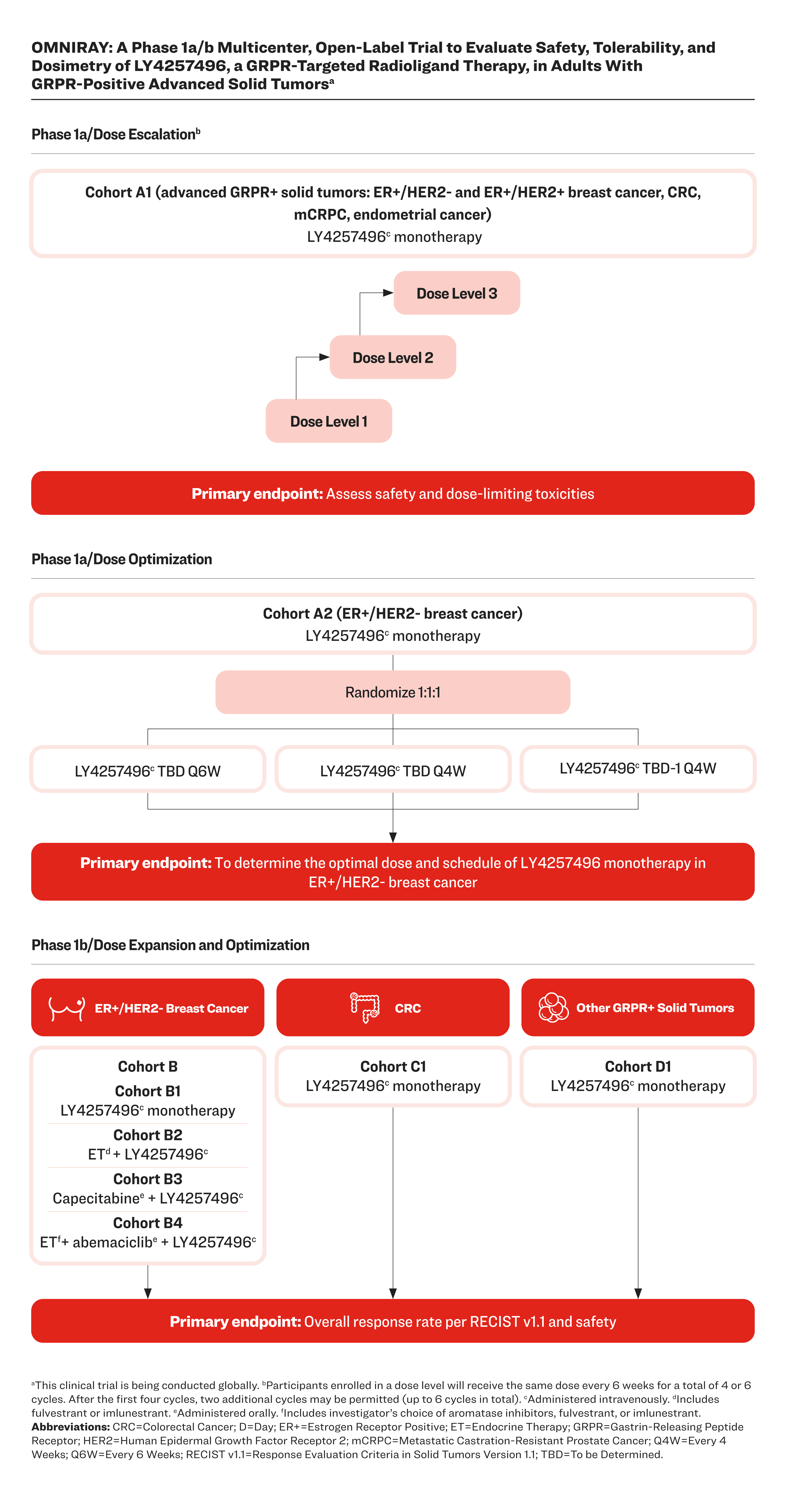
Key Inclusion Criteria
Key Exclusion Criteria
a
This clinical trial is being conducted globally.
b
Participants enrolled in a dose level will receive the same dose every 6 weeks for a total of 4 or 6 cycles. After the first four cycles, two additional cycles may be permitted (up to 6 cycles in total).
c
Administered intravenously.
d
Includes fulvestrant or imlunestrant.
e
Administered orally.
f
Includes investigator’s choice of aromatase inhibitors, fulvestrant, or imlunestrant.

